


Companion diagnostics (CDx) play a pivotal role in personalized medicine by enabling the identification of patients who are most likely to benefit from a specific therapeutic intervention. The development of these essential tools involves several key stages, each with unique challenges and complexities. In this article, we will discuss strategies for biomarker development and key considerations for early phase studies involving biomarkers for patient selection. We will also cover the regulatory requirements for use of a clinical trial assay (CTA) in early phase studies before the assay is ready for marketing authorization as a CDx.
Strategies for Biomarker Development
CDx development begins with biomarker discovery and validation. A biomarker can be any characteristic that indicates normal or pathogenic processes or predicts a response to an intervention. Biomarkers have diverse applications, from diagnosing disease and monitoring progression to assessing risk of disease and evaluating responses to therapeutic interventions. The assessed biomarker characteristic can be, for example, a single mutated gene associated with a disease state, an immune marker associated with therapeutic efficacy, or antibodies to a specific adeno-associated virus serotype viral vector.
When a biomarker is formally developed into a CDx, it becomes a device that provides essential information for the safe, effective use of a corresponding drug or biological product. CDx can be used to:
- Identify patients who are most likely to benefit from a particular therapeutic product
- Identify patients who are likely to be at increased risk for serious side effects resulting from treatment with a particular therapeutic product
- Monitor response to a particular therapeutic product for the purpose of adjusting treatment to achieve improved safety or effectiveness
Key Considerations for Early Phase Studies of Single-Gene Biomarkers
When developing a single-gene oncology biomarker, prior to enrolling patients, it is ideal to have a locked biomarker definition specifying which alteration(s) constitute a “biomarker positive” result, such as an exon 19 deletion in the EGFR gene. This means prior to initiation of a registrational study the validated biomarker should not change.
Before the biomarker is developed into a marketed CDx assay, it is clinically validated within the registrational study and the assay used for patient enrollment in this context is called a clinical trial assay. Optimally, the clinical trial assay will have been analytically validated within one central CLIA validated laboratory. However, when multiple local tests are used to enroll patients, it is critical to evaluate these tests for differences in cutoffs, analytical sensitivity, and accuracy. In such scenarios, best practices include ensuring that each test covers all alterations within the biomarker definition and collecting as much assay-specific information as possible per site to support equivalent performance claims.
If the biomarker definition is still in development in early phase studies, an initial clinical signal may be limited due to the prevalence of alterations. In this case, it is important to rely on preclinical data and published literature to support the biomarker definition.
Key Considerations for Early Phase Studies of Complex Signature Biomarkers
CDx may also involve complex signature biomarkers, which provide a continuum of numerical values that requires the establishment of a cutoff for treatment eligibility. The cutoff for a complex biomarker signature can be established through retrospective analysis of a completed therapeutic study, where such analysis reveals clinical efficacy for a subset of patients who may not have been evaluated for that signature upon enrollment. For many early phase studies where the cutoff is not known, applying an all-comers approach or performing a retrospective analysis to establish the cutoff for later phase studies is recommended because there may be differences in clinical efficacy based on expression levels. When the initial signature is established through retrospective analysis, separate patient populations will be needed for cutoff establishment and cutoff validation prior to using the validated cutoff in a registrational study.
Key Considerations for Early Phase Studies of Antibody Expression in Gene Therapy
Neutralizing antibody (NAb) assays which may be used to evaluate antibody levels in a patient’s sample prior to administration of gene therapy, are not as precise as traditional diagnostics. These assays may be either qualitative or semiquantitative. For qualitative assays, the cutoff can be the limit of detection or the signal at the minimum required dilution. For semiquantitative assays, the cutoff can be above the limit of detection and will be a specific titer tied to clinical efficacy. Each of these approaches has its pros and cons (see Figure 1).
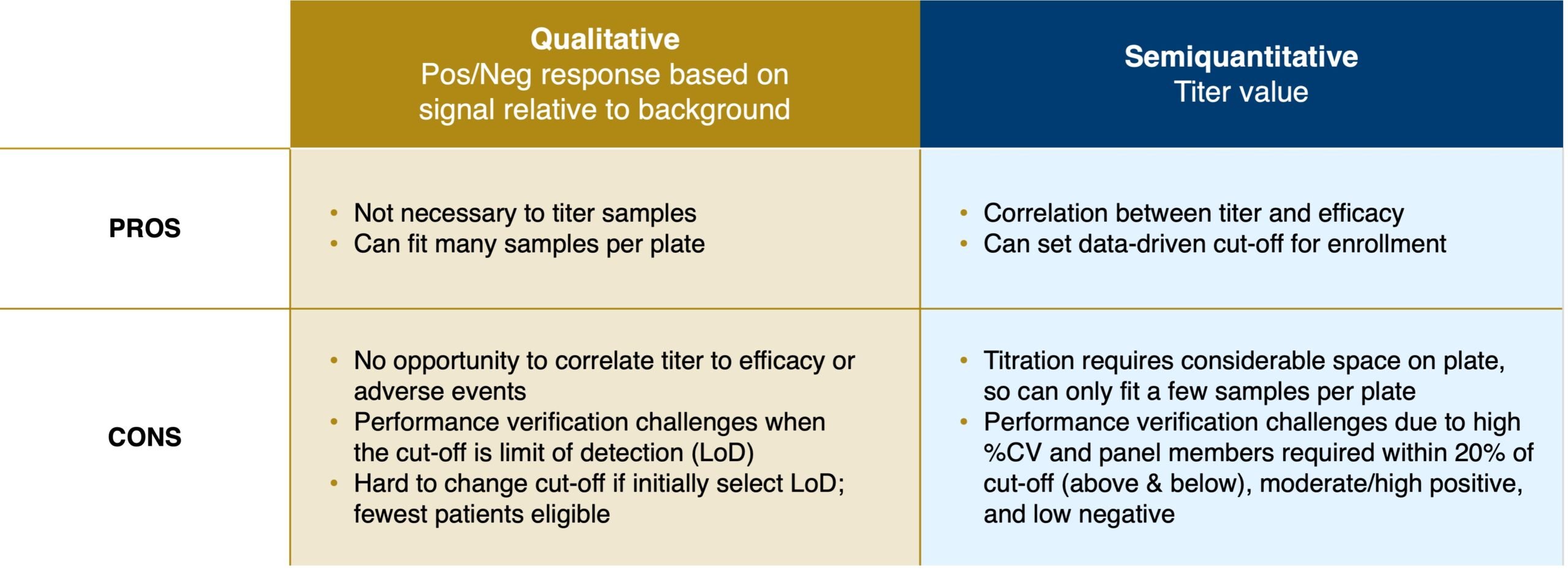
Figure 1. Qualitative vs Semiquantitative NAb assays
Regulatory Submissions for Clinical Trial Assays
Before an assay achieves marketing authorization as a CDx, the assay will need to be used in an investigational study to establish the clinical utility of the assay in a specific patient population. In this context, the assay is considered a clinical trial assay (CTA). Following the investigational device exemption (IDE) regulations, all devices used in clinical investigations must have an approved IDE, abbreviated IDE, or be exempt from the IDE regulations. The requirement for an IDE for a CTA is determined based on how the CTA is used in the context of the clinical study as well as the level of risk introduced by use of the CTA as part of the clinical trial (see Figure 2). An IDE allows a medical device that has not received marketing clearance or approval to be shipped for use in a clinical study without complying with other regulations of the Federal Food, Drug, and Cosmetic Act.
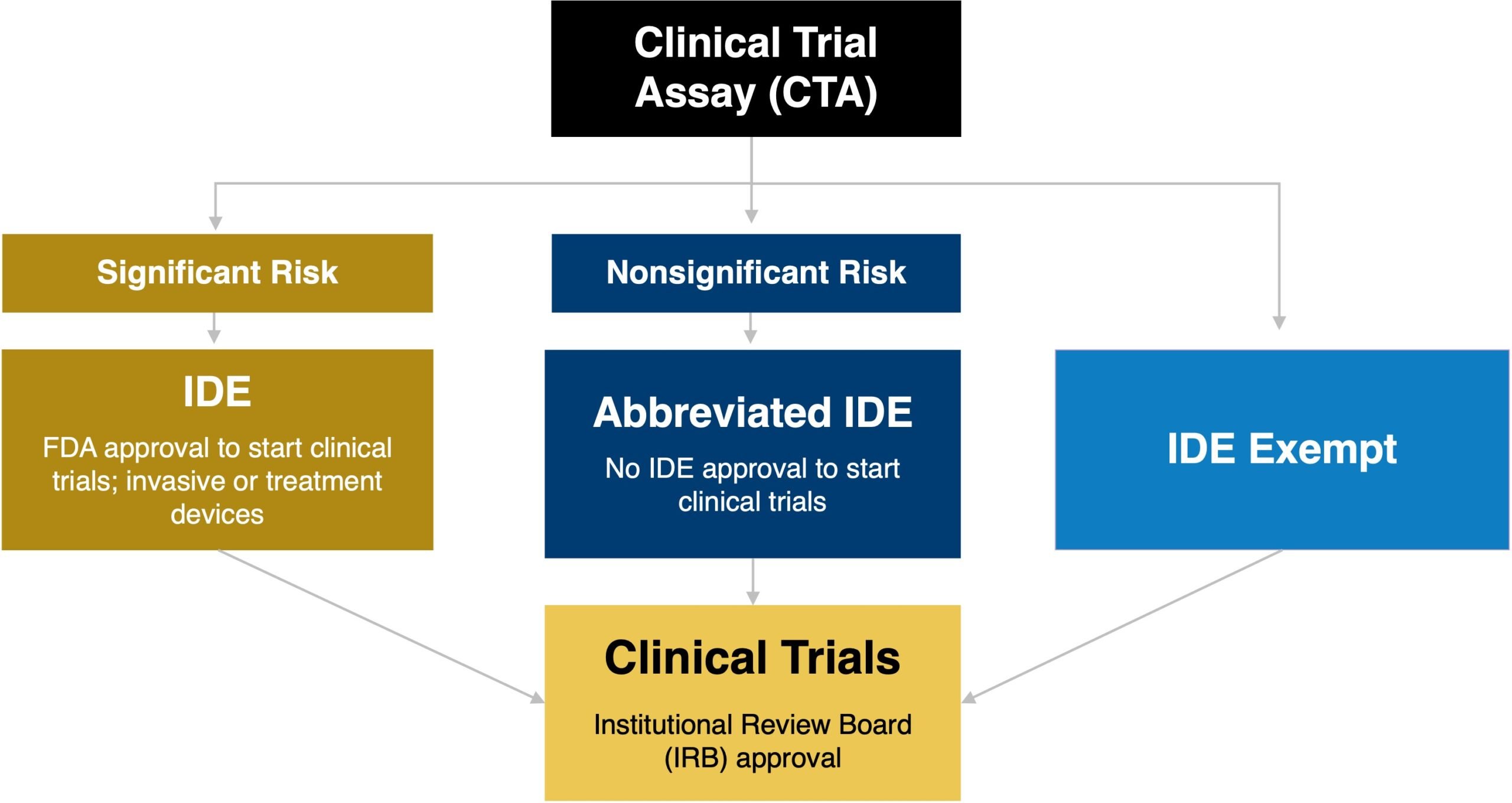
Figure 2. Overview of the Regulatory Requirements for the use of a Clinical Trial Assay based on the Risk Assessment
Key questions to help assess the level or risk presented by use of the device in the context of the clinical study include:
Study Considerations
- Are the test results used to determine whether the patient receives treatment?
- Would a false-positive result lead to the patient not receiving a known and effective therapy or standard of care (SOC)?
- How does the safety profile of the experimental therapeutic compare to the SOC?
- Have patients exhausted all SOC options?
Device Considerations
- Has the device been used in prior investigations with available safety data?
- Is the test being used prospectively or retrospectively?
- What sort of sample is required for the test, and does it involve an invasive procedure?
- If a fresh biopsy is required, is it a significant risk procedure such as a biopsy of the brain, lung, or pancreas?
If the CTA is used to determine who receives treatment, either via prospective stratification or prospective enrollment of only assay-positive patients, a study risk determination (SRD) Q-submission can be submitted to FDA to determine the risk associated with the use of the assay in the clinical trial. Based on the feedback from FDA the assay will be deemed NSR or SR which means an IDE will be required (Figure 3).
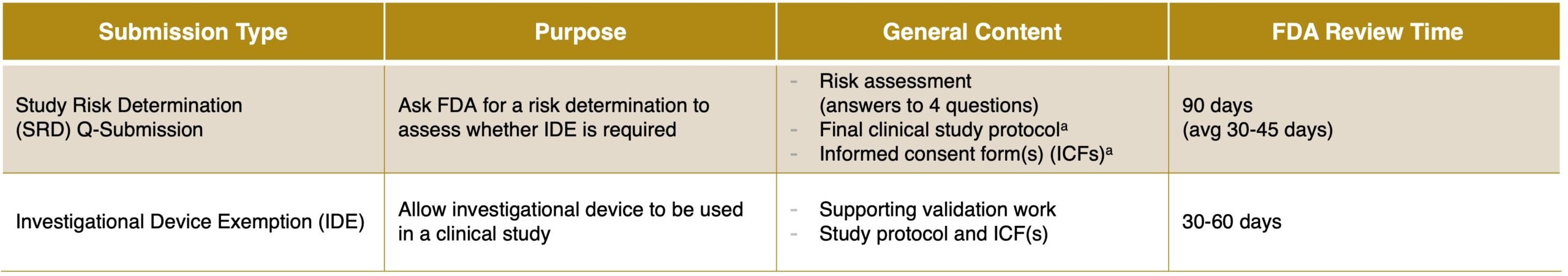
Figure 3. Summary of the SRD vs IDE for clinical trial assays
SRD Q-Submission: Content and Timing
Compared to an IDE, an SRD Q-Submission is a streamlined submission that unlike an IDE does not include the analytical validation data for the CTA. This approach can be favorable and preferred for the risk assessment if a strong argument can be made that use of the device in the context of the clinical study is low risk. The SRD submission includes:
- Background information on the study compound and disease state
- Summary of the investigational plan, including the informed consent forms and final clinical study protocol
- Intended use for the CTA and a description of the device
- Risk assessment that answers four questions from FDA draft guidance demonstrating non-significant risk
IDE Submission: Content and Timing
Compared to an SRD Q-submission, the content of an IDE is more comprehensive and includes the following:
- Background information on the study compound and disease state
- Summary of the investigational plan, including the informed consent forms and final clinical study protocol
- Information on prior studies involving the study compound and diagnostic
- A description of the methods, facilities, and controls used for the assay
- Device description with definition of biomarker
- Validation studies, including concordance data
For developers with an assay that has not yet been validated or questions about how to validate a CTA, it is highly recommended to do a pre-IDE Q-submission, which allows for feedback from the Center for Devices and Radiological Health (CDRH) on approximately 3-4 topics prior to submitting an IDE. Ideally, the pre-IDE Q-submission would include draft protocols, or at least detailed summaries of the proposed study designs. CDRH can provide feedback either in written form or via teleconference as to the adequacy of the proposed study designs to support an IDE application approval.
Key Takeaway
Effective development of a CDx requires early planning and disciplined execution beginning with biomarker identification and initial clinical investigations and extending through registrational studies and marketing approval. In the early phases of development, it is critical to establish and to validate cutoffs as well as understand the IDE regulatory requirements for use of a clinical trial assay for patient inclusion and exclusion. On the path of assay development to CDx marketing authorization, all assays will need to be used within an investigational study as a CTA to generate supporting clinical utility data to enable commercialization.
At Precision for Medicine, we bring together new technologies, regulatory and scientific expertise, and operational scale to accelerate companion diagnostic and therapeutic timelines. We provide comprehensive CDx solutions, from biomarker assay development, clinical trial enabling regulatory approvals, and clinical trial testing to data management services, regulatory strategy and submission development, liaison with regulatory bodies, and parallel therapeutic support.



